|
|
|
|
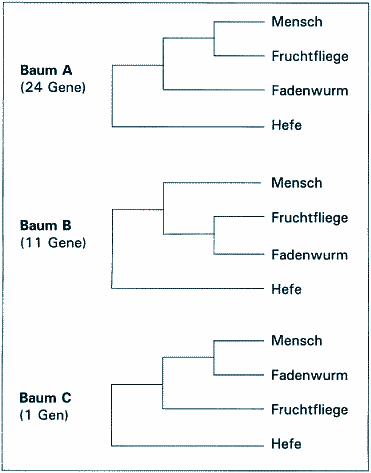
Es gab eine Zeit, da war es noch relativ einfach, sich ein Protein (z.B. das Cytochrom c) vorzunehmen, dessen Aminosäuresequenz bei verschiedenen Organismen zu bestimmen und aus den Daten einen Baum zu errechnen, der wunderbar mit der vermuteten Evolution der Lebewesen übereinstimmte, wenn man das richtige Protein und die richtigen Organismen ausgesucht hatte. Inzwischen beschäftigen sich Hunderte, ja Tausende von Arbeiten mit der Rekonstruktion von Verwandtschaftsbeziehungen auf der Basis von Makromolekül-Sequenzen (Proteine, RNS und DNS), so daß sich daraus mittlerweile ein eigenes Fachgebiet innerhalb der Systematik entwickelt hat. Aufgrund des technischen Fortschritts ist es sogar möglich geworden, ganze Genome, also das komplette Erbgut von Organismen, zu vergleichen. Auf jeder bis dato erreichten Ebene vergrößerten sich die Erwartungen an die Aussagekraft von noch mehr Daten. Die Situation hat sich jedoch im Gegenteil erheblich verkompliziert. Denn nahezu jedes Gen evolviert mit einer anderen Rate, hat andere Substitutionsmuster und -wahrscheinlichkeiten. Widersprüche auf breiter Front sind die Folge. Doch daran hat man sich inzwischen sogar gewöhnt. Immer spitzfindigere Verfahren werden entwickelt, mit deren Hilfe man diese Probleme in den Griff bekommen möchte. Der wissenschaftliche Nutzen dieser Anstrengungen ist erheblich: durch Sequenzähnlichkeiten kann z.B. auf ähnliche Funktionen geschlossen werden; Mechanismen der Genregulation, Prinzipien der Embryonalentwicklung oder Vorgänge im Genom (und damit auch Krankheitsursachen etc.) können wesentlich besser verstanden werden. Ein Beispiel dafür ist eine Studie (Mushegian et al. 1998), in der versucht wurde, durch solche Vergleiche Einsichten in die Funktion menschlicher Gene und Proteine zu gewinnen. Hierzu wurden 36 Proteine von Mensch, Fruchtfliege, einem Fadenwurm und einer Hefe verglichen (die Daten stammen aus Genomsequenzierungsprojekten). Drei prinzipielle Möglichkeiten, aus diesen vier Arten einen Baum (Kladogramm) zu konstruieren, existieren - vorausgesetzt, man setzt die Hefe an die Wurzel des Baumes (Abb. 1). Nun sprachen 24 Proteine für Möglichkeit A, 11 für B und ein Protein für C. Möglichkeit A entspricht der konventionellen Interpretation der Stammesgeschichte. Handelte es sich dann bei den anderen Ergebnissen nur um "störendes Rauschen"? Eine genauere Betrachtung der Daten zeigte, daß die homogen, also mit ähnlicher Rate evolvierenden Proteine, Baum B ergaben, heterogen evolvierende dagegen Baum A. Dies würde bedeuten, daß ungleiche Evolutionsraten (und nicht Abstammungsverhältnisse) dafür verantwortlich waren, daß die Mehrzahl der Proteine Baum A unterstützte. Welcher ist nun der "richtige"? |
|
Auch die Erbinformation der Mitochondrien ist heute schon für zahlreiche Organismen komplett bekannt, und immer mehr Untersuchungen benutzen diese Daten für Rekonstruktionen der Stammesgeschichte. Ein jüngeres Beispiel dafür ist eine Studie über Fische (Rasmussen & Arnason 1999). Zum Basiswissen der Lehrbücher über Evolution gehört, daß Knorpelfische (z.B. Haie) älter sind als Knochenfische (fast alle übrigen, heute noch lebenden Fische) und ihnen in vieler Hinsicht nur oberflächlich gleichen. Der Vergleich des kompletten mitochondrialen Genoms läßt jedoch einen Knorpelfisch mitten in den Knochenfischen (inklusive Neunauge, Lungenfisch und Quastenflosser) auftauchen. Hierbei ist zu beachten, daß ein wesentlicher Aspekt solcher Analysen die Auswahl eines möglichst nahe verwandten Organismus ist, der außerhalb der untersuchten Arten steht, und mit dessen Einbeziehung die Wurzel des Baumes gefunden werden soll - der subjektivste Teil der Analyse. Vielfach hängt die Topologie eines Verwandtschaftsdiagramms von der Wahl dieser so genannten Außengruppe ab (Takezaki & Gojobori 1999) sowie von der Methode, die benutzt wurde, um den Baum zu erstellen. Auch "biologisch falsche", d.h. aller sonstigen Evidenz widersprechende und offenbar unsinnige Phylogenien können dabei hohe statistische Absicherung erfahren.
Mögliche Erklärungen für solche Ungereimtheiten und Diskrepanzen sind z.B. Änderungen in der Aminosäurezusammensetzung oder der Austauschgeschwindigkeit in verschiedenen Evolutionslinien, mehrfach voneinander unabhängiges Auftauchen der gleichen Aminosäuren, Variation der Evolutionsraten an verschiedenen Positionen oder unterschiedliche Austauschwahrscheinlichkeiten für bestimmte Aminosäuren. Diese Möglichkeiten müssen im Einzelfall nachgeprüft werden. Manche Unstimmigkeit kann durch detailliertere Analysen aufgelöst werden; dennoch ist die Gefahr von Zirkelschlüssen nicht gebannt: Keiner weiß, wie die (molekulare) Evolution tatsächlich abgelaufen sein mag, daher sind die Modelle letztlich kaum verifizierbar. Die Übereinstimmung mit morphologischen Befunden ist zwar eine gewisse Rückversicherung, aber keine wirkliche Hilfe, wenn deren Ergebnisse mit molekularen Methoden überprüft werden sollen. Eine annahmenfreie Rekonstruktion von Evolution ist also auch in der molekularen Systematik nicht möglich. Bei all diesen Überlegungen wird von der Homologie der untersuchten Makromoleküle ausgegangen (die nach anerkannten Regeln geprüft wird), also von der Annahme einer durch gemeinsame Vererbung entstandenen Ähnlichkeit. Dahinter steht die drastische, aber allgemein akzeptierte Grundannahme, daß es Evolution im Sinne von Makroevolution wirklich gegeben hat. Geht man jedoch vom Grundtypmodell aus (Scherer 1993), so existiert echte (phylogenetisch bedingte) Homologie nur für Gene/Proteine innerhalb von Grundtypen. Auch hier ist die Rekonstruktion von Verwandtschaft mitunter nicht einfach (Fehrer 1996); sie wirft vielmehr zusätzliche Probleme auf, wenn mit Hybridisierungen und dadurch entstandenen retikulaten (netzförmigen) Beziehungen oder mit jungen, noch unvollständig getrennten Linien gerechnet werden muß. Dennoch hat die Beschränkung phylogenetischer Rekonstruktionen auf Grundtypen einen unschätzbaren Vorteil: man arbeitet tatsächlich mit vergleichbaren Merkmalskomplexen. Daraus läßt sich eine gewagte Hypothese formulieren: Das immense Anwachsen des Datenmaterials könnte auf Grundtypebene zu mehr Klärung der Beziehungen beitragen; im Fall von Vergleichen außerhalb der Grundtypen ist möglicherweise mit dem umgekehrten Effekt zu rechnen: je mehr Daten, desto mehr Widersprüche. Warten wir's ab. |

|